以下依据药监局(NMPA)药械组合法规(特别是2021年第52号通告及相关附件),梳理地药械组合产品属性界定的程序、材料要求及操作要点:
一、属性界定核心原则
1.定义
药械组合产品指由药品与医疗器械共同组成,并作为一个单一实体生产的医疗产品。属性界定依据其首要作用方式(药品或医疗器械作用为主)确定注册路径。
2.界定必要性
若无法自行确定产品属性,必须在注册申报前向NMPA医疗器械标准管理中心(标管中心)申请属性界定。
二、药械组合产品属性界定程序
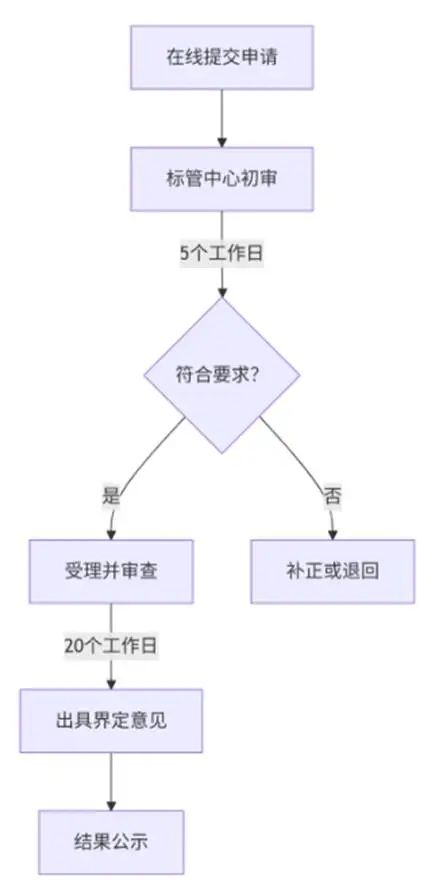
1. 申请流程
2. 材料要求
需上传以下材料(均需中文,外文资料需附原文):
(1)申请表:在线填写并签字签章后扫描上传;进口产品:填写国内代理人信息(机构名称、地址、联系人等)。
(2)支持性材料:
产品描述(名称、成分、组合方式、预期用途、接触部位/时间、示意图、实物照片等);
作用机制及相关验证资料(如体外实验、文献);
拟使用说明书(或用户手册);
组成成分来源说明;
属性界定建议及论证资料(包括首要作用方式判定及依据);
相关产品监管情况(如国内外已上市类似产品信息,进口产品提交境外上市的相关资料);
其他与属性确定相关的资料。
关键时限要求
环节 | 时限 | 说明 |
初审 | 5个工作日 | 资料不完整则一次性通知补正 |
审查与意见出具 | 20个工作日 | 专家论证时间不计入(需提前说明) |
补正资料提交 | 60个工作日 | 逾期未补或补正不符则退回申请 |
复审申请 | 收到结果后10个工作日 | 仅限原申请资料复审 |
三、结果应用
1.注册申报
申请人根据最终属性界定结果,向国家药监局申报药品或医疗器械注册,并在申请表中注明“药械组合产品”。
2.审评机制
注:双方分别出具安全性、有效性及质量可控性审评报告,牵头单位汇总后提交审批。




